ThunderBio will provide human and mouse pre-built reference and two sets example data upon request.
The demo_data fold contains two directories, human and mouse. In each directory, user could invoke
the pipeline by running command below. Please note the reference index has to be placed in the
same parent folder of the demo_data.
## Human Data Testing with conda or docker
cd demo_data/human
starscope run --input sampleList.csv --config thunderbio_human_conda.config
starscope run --input sampleList.csv --config thunderbio_human_docker.config
## Mouse Data Testing with conda or docekr
cd demo_data/mouse
Updated script for saturation calculation, and included UMI and gene hist data for {preseqR} prediction
Now a tsv summary file will be outputted in the report process
StarScope v1.1.3
Added --outBAMsortingBinsN 300 option to STARsolo process to solve the RAM issue when sorting a very large BAM file, refer to STAR #870
StarScope v1.1.2
Use soft link instead of cat when there is only one fastq file
Added support for VDJ subworkflow to only analyze BCR or TCR dataset
Added support for publishing saturation json file
StarScope v1.1.1
Honer the memory option in the report process
Added option for publishing starsolo BAM output
Added option to enable conda env for nextflow 22.10, and the nextflow version was fixed to 22.04
StarScope v1.1.0
Added VDJ workflow (GEX+VDJ)
Removed time limit for process resources
Modified the margin of the tables in the 3’-scRNA-Seq report
Adjusted knee plot to fixed size and center positioned
StarScope v1.0.0
Added pipeline running information to the reports
Fixed issue for saturation calculation script when there is no whitelist provided
Output a full list of DEGs
StarScope v0.0.9
Updated STAR to v2.7.10a in both the docker image and the conda environment
Adapt the pipeline to generate report for starsolo --soloMultiMappers option, only “Unique” and “EM” are supported by now.
Changed the default parameter for --soloFeatures to “GeneFull”, which includes both exon and intron reads
StarScope v0.0.8
Fixed the issue that RunPCA uses up all cores and consumes too much memory (Seurat #3991)
Changed h5file format to H5Seurat
Disabled sass cache in rmd report
Changed future package strategy to multisession to avoid fork errors
StarScope v0.0.7
Raw gene/barcode count table will be stored in a loom file and published for subsequent analysis
Mapping metrics will be written to a json file
Added support for 5’ RNA-seq library
StarScope v0.0.6
Now the BAM output will not by published
Added support for counting intron reads
Added check_version command, to print all software versions including docker or conda engine information
Fixed STAR to 2.7.9a and samtools to 1.15
Fixed issue that no report generated if too few cells detected
StarScope v0.0.5
Fixed issue that no report generated if there are too few cells left after filtering by nFeature >= 200
StarScope v0.0.3, v0.0.4
Set thunderbio data running parameters as defaults instead of 10x parameters
Added whitelists for thunderbio V2, 10x V2 and V3
Fixed nextflow default path issue
Fixed conda env name issue
Implemented docker container running environment
Added a new script for user to generate 10x compatible STAR reference
StarScope v0.0.2
Implemented mkref command
Added output section for README
StarScope v0.0.1
Implemented run command
Added running env checking procedure
Subsections of StarScope
Chapter 1
Basics
Instruction of the workflows.
Subsections of Basics
Installation
Requirements
Java 11 or higher
Nextflow
Conda/miniconda
Docker
Java
Nextflow will need java 11 or higher to be installed. The recommanded way to install
java is through SDKMAN. Please use the command below:
Install SDKMAN:
curl -s https://get.sdkman.io | bash
Open a new terminal and install Java
sdk install java 17.0.10-tem
Check java installation and comfirm it’s version
java -version
Nextflow
Nextflow binary was already included in the StarScope directory. User also could download binary
from nextflow’s github release page.
By default, starscope will invoke the nextflow executable stored in the same directory, user
could add both of the two executables to $PATH (e.g. ~/.local/bin)
Confirm that nextflow runs properly with the command below (require network access to github):
NXF_VER=23.10.1 nextflow run hello
The output will be:
N E X T F L O W ~ version 22.04.5
Launching `https://github.com/nextflow-io/hello` [distraught_ride] DSL2 - revision: 4eab81bd42 [master]
executor > local (4)
[92/5fbfca] process > sayHello (4) [100%] 4 of 4 ✔
Bonjour world!
Hello world!
Ciao world!
Hola world!
Conda/miniconda
Install Conda
We usually use miniforge’s conda distro, user also
could install via conda official installer, or use mamba directly, which is much faster.
Or extract environment from archive distributed by ThunderBio with conda-pack
# Unpack environment into directory `starscope_env`$ mkdir -p starscope_env
$ tar -xzf starscope_env.tar.gz -C starscope_env
# Activate the environment. This adds `starscope_env/bin` to your path$ source starscope_env/bin/activate
# Cleanup prefixes from in the active environment.# Note that this command can also be run without activating the environment# as long as some version of Python is already installed on the machine.(starscope_env) $ conda-unpack
# deactivete env$ source starscope_env/bin/deactivate
Docker
Using docker is much easier to integrate the workflow to large infrastructure
like cloud platforms or HPC, thus is recommended. To install:
curl -fsSL https://get.docker.com -o get-docker.sh
sh get-docker.sh
To use docker command without sudo, add your account to docker group:
sudo usermod -aG docker $(whoami)
Then log out and login again for the changes to take effect.
After invoking the pipeline, nextflow will report the progress
to stdout, with each row representing a process.
N E X T F L O W ~ version 23.10.1
Launching `/thunderData/pipeline/starscope/scRNA-seq/main.nf` [adoring_ekeblad] DSL2 - revision: 8e27902b23
executor > slurm (9)
[e0/1d00d4] process > scRNAseq:CAT_FASTQ (human_test) [100%] 1 of 1 ✔
[37/8c0795] process > scRNAseq:TRIM_FASTQ (human_test) [100%] 1 of 1 ✔
[20/1edf9b] process > scRNAseq:MULTIQC (human_test) [100%] 1 of 1 ✔
[5a/e0becc] process > scRNAseq:STARSOLO (human_test) [100%] 1 of 1 ✔
[02/15a3b1] process > scRNAseq:CHECK_SATURATION (human_test) [100%] 1 of 1 ✔
[09/e25428] process > scRNAseq:GET_VERSIONS (get_versions) [100%] 1 of 1 ✔
[48/703c20] process > scRNAseq:FEATURESTATS (human_test) [100%] 1 of 1 ✔
[79/cd2784] process > scRNAseq:GENECOVERAGE (human_test) [100%] 1 of 1 ✔
[e6/808adf] process > scRNAseq:REPORT (human_test) [100%] 1 of 1 ✔
Completed at: 09-May-2024 09:07:55
Duration : 25m 9s
CPU hours : 3.7
Succeeded : 9
When encountering any error, nextflow will interrupt running and
print error message to stderr directly.
User could also check the error message from running log file .nextflow.log
$ head .nextflow.log
May-09 08:42:37.523 [main] DEBUG nextflow.cli.Launcher - $> nextflow run /thunderData/pipeline/starscope/scRNA-seq -c /thunderData/pipeline/nf_scRNAseq_config/latest/thunderbio_human_config --input sampleList.csv
May-09 08:42:37.924 [main] INFO nextflow.cli.CmdRun - N E X T F L O W ~ version 23.10.1
May-09 08:42:38.096 [main] DEBUG nextflow.plugin.PluginsFacade - Setting up plugin manager > mode=prod;embedded=false; plugins-dir=/home/xzx/.nextflow/plugins; core-plugins: nf-amazon@2.1.4,nf-azure@1.3.3,nf-cloudcache@0.3.0,nf-codecommit@0.1.5,nf-console@1.0.6,nf-ga4gh@1.1.0,nf-google@1.8.3,nf-tower@1.6.3,nf-wave@1.0.1
May-09 08:42:38.147 [main] INFO o.pf4j.DefaultPluginStatusProvider - Enabled plugins: []May-09 08:42:38.150 [main] INFO o.pf4j.DefaultPluginStatusProvider - Disabled plugins: []May-09 08:42:38.163 [main] INFO org.pf4j.DefaultPluginManager - PF4J version 3.4.1 in 'deployment' mode
May-09 08:42:38.234 [main] INFO org.pf4j.AbstractPluginManager - No plugins
May-09 08:42:42.225 [main] DEBUG nextflow.config.ConfigBuilder - Found config base: /thunderData/pipeline/starscope/scRNA-seq/nextflow.config
May-09 08:42:42.231 [main] DEBUG nextflow.config.ConfigBuilder - User config file: /thunderData/pipeline/nf_scRNAseq_config/latest/thunderbio_human_config_v2
May-09 08:42:42.233 [main] DEBUG nextflow.config.ConfigBuilder - Parsing config file: /thunderData/pipeline/starscope/scRNA-seq/nextflow.config
Nextflow Log CLI
After each invokation, the pipeline running information could be retrieved by nextflow log
command, and user could check the RUN NAME, STATUS and SESSION ID from the command output.
$ nextflow log
TIMESTAMP DURATION RUN NAME STATUS REVISION ID SESSION ID COMMAND
2024-05-09 08:42:44 25m 12s adoring_ekeblad OK 8e27902b23 8670925f-ce5a-4f7a-b327-a98b288e6aa6 nextflow run /thunderData/pipeline/starscope/scRNA-seq -c /thunderData/pipeline/nf_scRNAseq_config/latest/thunderbio_human_config --input sampleList.csv
Work Dir and Intermediate Files
Each task of the process will be conducted in a sub-directory of the workDir set in
nextflow configuration file. By default, StarScope set this to work folder
under project running directory. To confirm each tasks’ working directory, user
will have to check the task hash id with command below. The adoring_ekeblad is
the RUN NAME from nextflow log output.
One of the core features of Nextflow is the ability to cache task executions and re-use
them in subsequent runs to minimize duplicate work. Resumability is useful both for recovering
from errors and for iteratively developing a pipeline.
It is similar to checkpointing,
a common practice used by HPC applications.
To resume from previous run, please use the command below after entering the project running directory:
By default, each run of the pipeline will generate
three tracing files in results/pipeline_info/, check
nextflow document for details.
execution_trace_<timeStamp>.txt
execution_timeline_<timeStamp>.html
execution_report_<timeStamp>.html
Trace Report
Nextflow generates an execution tracing tsv file with
valuable details on each process, including submission time,
start time, completion time, CPU usage,
and memory consumption.
Nextflow can also create an HTML timeline report for all pipeline processes.
See example below:
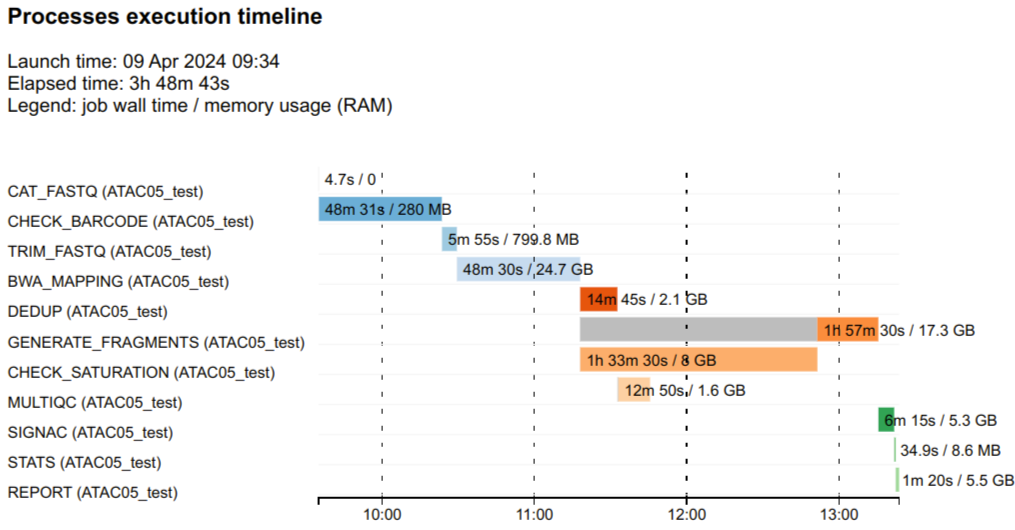
Each bar represents a single process run, the length of the bar represents
the duration time (wall-time). Colored part of the bar indicates real
processing time, grey part represents scheduling wait time.
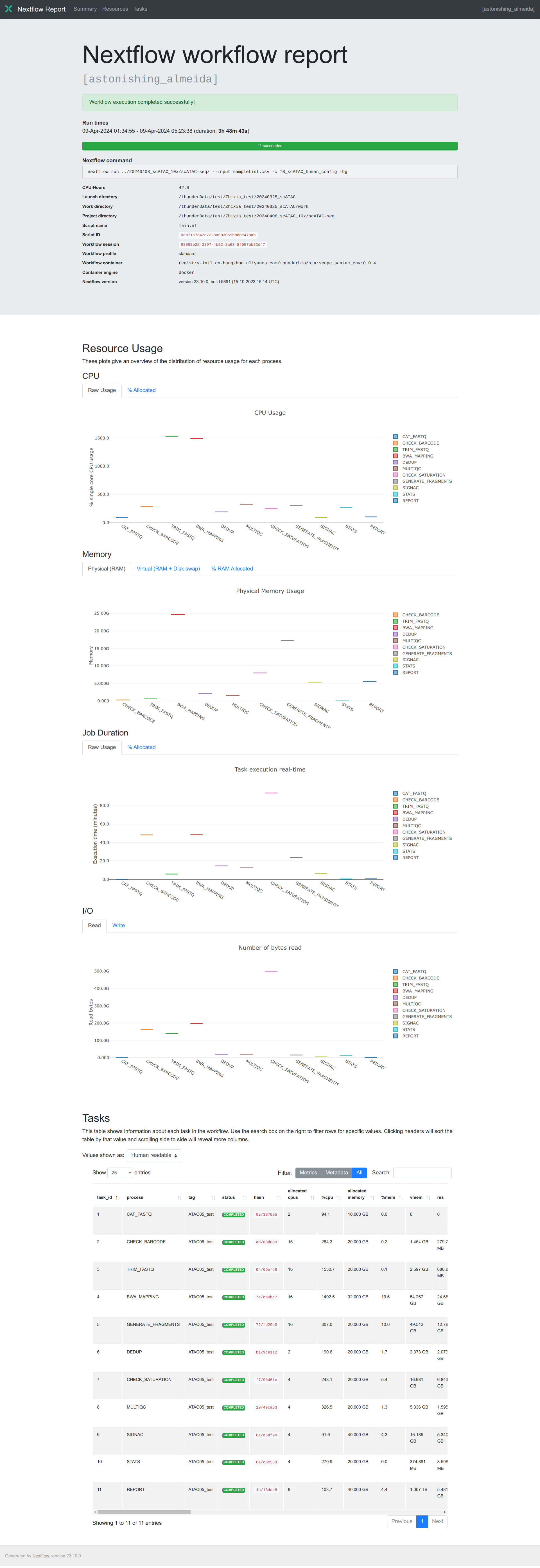
Execution report
The execution report is more concise, which logs running information in
the summary section, summarizes task resource usage
using plotly.js and collects task metrics like
trace report with more terms in a table.
Chapter 2
scRNA-seq Workflow
Subsections of scRNA-seq Workflow
Invoke with StarScope
Input
The input sample list file is a simple csv with four columns.
The first column sample indicates sample IDs, and multiple fastq
files with the same sample ID will be concatenated before further
processing (e.g. two pairs of human_pbmc_s1 fastq files will be
cat to a single pair). Multiple samples in one single sample list
will be submitted parallelly and processed asynchronously. The
fourth column indicates expected number of cells used for
starsolo--soloCellFilter parameter.
All samples in the sampleList will use the same options. Therefore, a combination of samples from different species is not supported.
Command
Invoke with config file
Due to the abundance of processes and options, we suggest utilizing a custom
configuration file when invoking the pipeline. This allows for greater
control over resource management. ThunderBio has released two versions of the
chemistry, each with a distinct barcode structure. Please refer to the
corresponding configuration examples provided below.
params{genomeDir="/refdata/human/starsolo/"genomeGTF="/refdata/human/refdata-gex-GRCh38-2020-A/genes/genes.gtf"whitelist="starscope/whitelist/V2_barcode_seq_210407_concat.txt"trimLength=50soloType="CB_UMI_Simple"soloCBstart=1soloCBlen=29soloUMIstart=30soloUMIlen=10publishSaturation=true}// uncomment line below if using slurm, see https://www.nextflow.io/docs/latest/executor.html
// process.executor = 'slurm'
// uncomment below chunk if using conda
// process.conda = "/home/xzx/Tools/mambaforge/envs/starscope_scRNAseq_env"
// conda.enabled = true
// docker setting, comment chunk below if using conda
process.container="registry-intl.cn-hangzhou.aliyuncs.com/thunderbio/starscope_scrnaseq_env:1.2.5"docker.enabled=truedocker.userEmulation=truedocker.runOptions='--init-u$(id-u):$(id-g)$(opt="";forgroupin$(id-G);doopt=$opt" --group-add $group";done;echo$opt)'// Resouces for each process
process{withLabel:process_high{cpus=16memory=40.GB}withLabel:process_medium{cpus=4memory=20.GB}withLabel:process_low{cpus=4memory=20.GB}withName:CHECK_SATURATION{cpus=4memory=10.GB}withName:CAT_FASTQ{cpus=2memory=4.GB}withName:TRIM_FASTQ{cpus=12memory=20.GB}withName:MULTIQC{cpus=4memory=10.GB}withName:STARSOLO{cpus=16memory=40.GB}withName:REPORT{cpus=4memory=40.GB}withName:FEATURESTATS{cpus=2memory=8.GB}withName:GENECOVERAGE{cpus=8memory=10.GB}}
params{genomeDir="/refdata/human/starsolo/"genomeGTF="/refdata/human/refdata-gex-GRCh38-2020-A/genes/genes.gtf"whitelist="3M-february-2018.txt"trimLength=28soloType="CB_UMI_Simple"soloCBstart=1soloCBlen=16soloUMIstart=17soloUMIlen=12publishSaturation=true}// uncomment line below if using slurm, see https://www.nextflow.io/docs/latest/executor.html
// process.executor = 'slurm'
// uncomment below chunk if using conda
// process.conda = "/home/xzx/Tools/mambaforge/envs/starscope_scRNAseq_env"
// conda.enabled = true
// docker setting, comment chunk below if using conda
process.container="registry-intl.cn-hangzhou.aliyuncs.com/thunderbio/starscope_scrnaseq_env:1.2.5"docker.enabled=truedocker.userEmulation=truedocker.runOptions='--init-u$(id-u):$(id-g)$(opt="";forgroupin$(id-G);doopt=$opt" --group-add $group";done;echo$opt)'// Resouces for each process
process{withLabel:process_high{cpus=16memory=40.GB}withLabel:process_medium{cpus=4memory=20.GB}withLabel:process_low{cpus=4memory=20.GB}withLabel:process_high{cpus=32memory=32.GB}withLabel:process_medium{cpus=32memory=20.GB}withLabel:process_low{cpus=4memory=20.GB}withName:CHECK_SATURATION{cpus=4memory=10.GB}withName:CAT_FASTQ{cpus=2memory=4.GB}withName:TRIM_FASTQ{cpus=12memory=20.GB}withName:MULTIQC{cpus=4memory=10.GB}withName:STARSOLO{cpus=16memory=40.GB}withName:REPORT{cpus=4memory=40.GB}withName:FEATURESTATS{cpus=2memory=8.GB}withName:GENECOVERAGE{cpus=8memory=10.GB}}
Invoke with command line options
To invoke scRNA-seq pipeline with conda environment:
soloType None
string(s): type of single-cell RNA-seq
CB_UMI_Simple ... (a.k.a. Droplet) one UMI and one Cell Barcode of fixed length in read2, e.g. Drop-seq and 10X Chromium.
CB_UMI_Complex ... one UMI of fixed length, but multiple Cell Barcodes of varying length, as well as adapters sequences are allowed in read2 only, e.g. inDrop.
CB_samTagOut (Not supported) ... output Cell Barcode as CR and/or CB SAm tag. No UMI counting. --readFilesIn cDNA_read1 [cDNA_read2 if paired-end] CellBarcode_read . Requires --outSAMtype BAM Unsorted [and/or SortedByCoordinate] (Not supported by StarScope for now)
SmartSeq (Not supported) ... Smart-seq: each cell in a separate FASTQ (paired- or single-end), barcodes are corresponding read-groups, no UMI sequences, alignments deduplicated according to alignment start and end (after extending soft-clipped bases) (Not supported by StarScope for now)
soloCBstart 1
int>0: cell barcode start base
soloCBlen 16
int>0: cell barcode length
soloUMIstart 17
int>0: UMI start base
soloUMIlen 10
int>0: UMI length
soloCBposition -
strings(s) position of Cell Barcode(s) on the barcode read.
Presently only works with --soloType CB_UMI_Complex, and barcodes are assumed to be on Read2.
Format for each barcode: startAnchor_startPosition_endAnchor_endPosition
start(end)Anchor defines the Anchor Base for the CB: 0: read start; 1: read end; 2: adapter start; 3: adapter end
start(end)Position is the 0-based position with of the CB start(end) with respect to the Anchor Base
String for different barcodes are separated by space.
Example: inDrop (Zilionis et al, Nat. Protocols, 2017):
--soloCBposition 0_0_2_-1 3_1_3_8
soloUMIposition -
string position of the UMI on the barcode read, same as soloCBposition
Example: inDrop (Zilionis et al, Nat. Protocols, 2017):
--soloCBposition 3_9_3_14
soloAdapterSequence -
string: adapter sequence to anchor barcodes.
soloCellFilter CellRanger2.2 3000 0.99 10
string(s): cell filtering type and parameters
None ... do not output filtered cells
TopCells ... only report top cells by UMI count, followed by the exact number of cells
CellRanger2.2 ... simple filtering of CellRanger 2.2.
Can be followed by numbers: number of expected cells, robust maximum percentile for UMI count, maximum to minimum ratio for UMI count
The harcoded values are from CellRanger: nExpectedCells=3000; maxPercentile=0.99; maxMinRatio=10
EmptyDrops_CR ... EmptyDrops filtering in CellRanger flavor. Please cite the original EmptyDrops paper: A.T.L Lun et al, Genome Biology, 20, 63 (2019): https://genomebiology.biomedcentral.com/articles/10.1186/s13059-019-1662-y
Can be followed by 10 numeric parameters: nExpectedCells maxPercentile maxMinRatio indMin indMax umiMin umiMinFracMedian candMaxN FDR simN
The harcoded values are from CellRanger: 3000 0.99 10 45000 90000 500 0.01 20000 0.01 10000
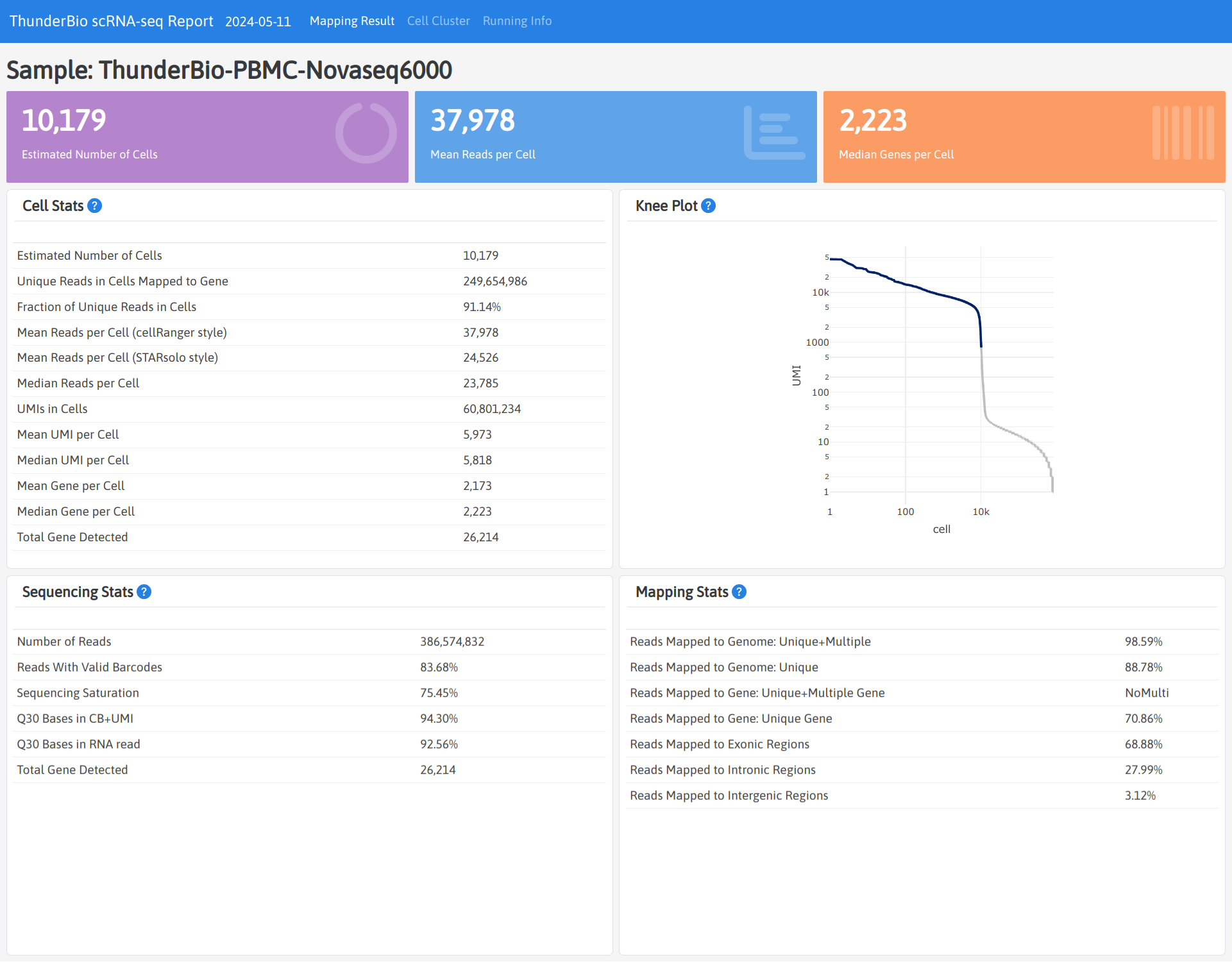
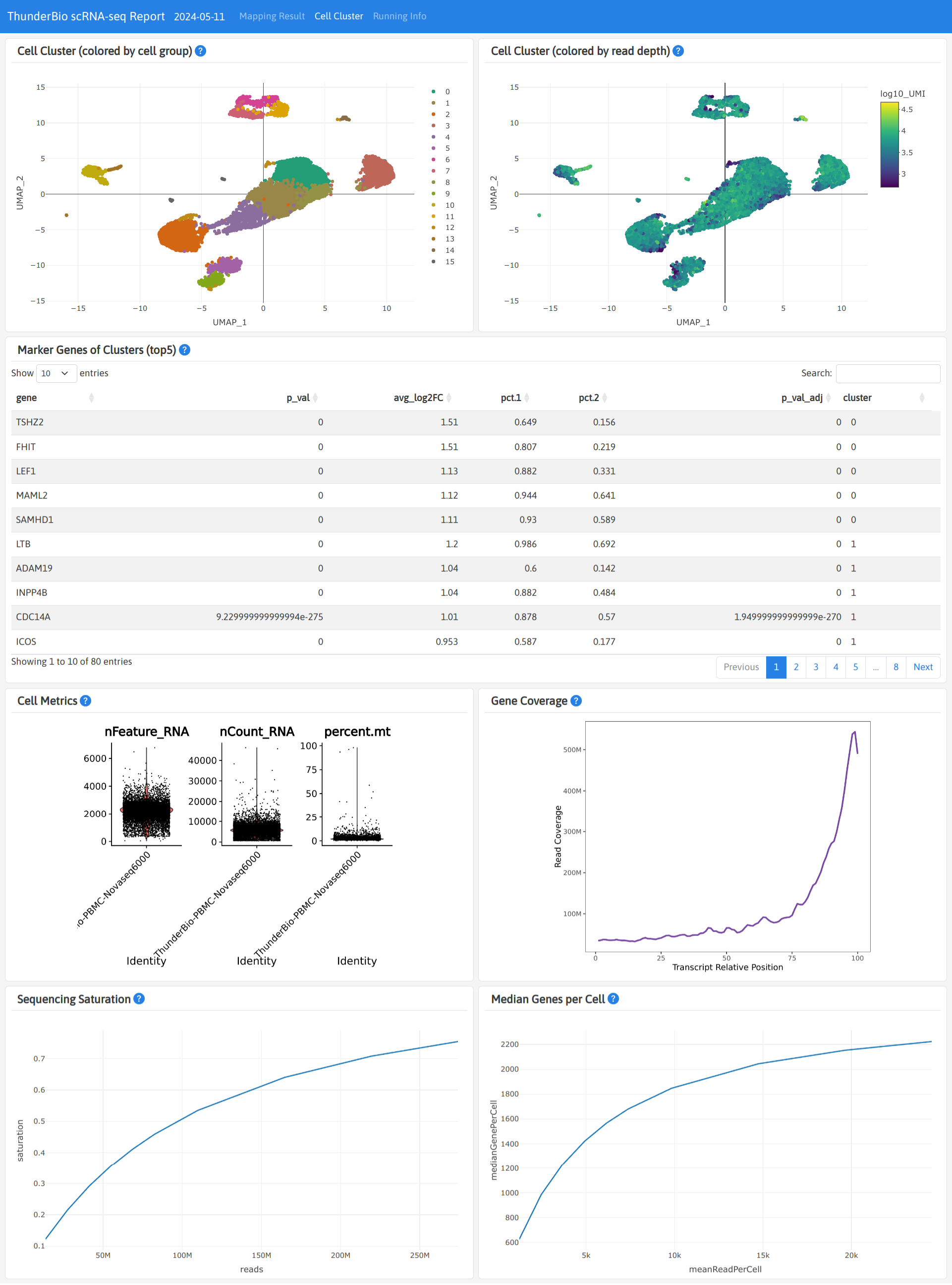
Outputs
Each sample will have a separated result folder named with sample ID. The sub-directory final
contains most of the result files, including html report, gene expression matrix (filtered:
contains cell associated barcodes only; raw: contains all barcodes).
The pipeline_info directory contains statistics of the pipeline running resources.
By default, the intermediate files will be written to sub-directory of
work under the pipeline running directory, please feel free to
remove it after all the processes finished successfully.
Make Reference
To build a genome reference index, StarScope included a subcommand
mkref to help user achieve this via nextflow run.
The output files are genome.fa and genes.gtf in the working directory.
Chapter 3
VDJ Workflow
Chapter homepage for VDJ immune repertoire analysis module.
Subsections of VDJ Workflow
Invoke with StarScope
Input
The input sample list file is csv file with five columns: sample,
fastq_1, fastq_2, feature_types and expected_cells. The
fourth column represents library types: GEX, VDJ-T or VDJ-B.
A sample with only VDJ-T or VDJ-B library is supported, but
all the samples must have at least one VDJ library. Please
use scRNA-seq workflow if you only have GEX library.
The first column sample indicates sample IDs, and multiple fastq
files with the same sample ID will be concatenated before further
processing (e.g. two pairs of human_pbmc_s1 fastq files will be
cat to a single pair). Multiple samples in one single sample list
will be submitted parallelly and processed asynchronously. The
fourth column indicates expected number of cells used for
starsolo--soloCellFilter parameter.
All samples in the sampleList will use the same options. Therefore, a combination of samples from different species is not supported.
Command
Invoke with config file
Due to the abundance of processes and options, we suggest utilizing a custom
configuration file when invoking the pipeline. This allows for greater
control over resource management. ThunderBio has released two versions of the
chemistry, each with a distinct barcode structure. Please refer to the
corresponding configuration examples provided below.
params{genomeDir="/refdata/human/starsolo/"genomeGTF="/refdata/human/refdata-gex-GRCh38-2020-A/genes/genes.gtf"whitelist="/starscope/whitelist/TB_v3_20240429.BC1.tsv /starscope/whitelist/TB_v3_20240429.BC2.tsv /starscope/whitelist/TB_v3_20240429.BC3.tsv"soloType="CB_UMI_Complex"trimLength=50soloAdapterSequence="NNNNNNNNNGTGANNNNNNNNNGACANNNNNNNNNNNNNNNNN"soloCBposition="2_0_2_8 2_13_2_21 2_26_2_34"soloUMIposition="2_35_2_42"soloCBmatchWLtype="1MM"publishSaturation=true// 5' VDJ specific params
// trust4 reference
trust4_vdj_refGenome_fasta="/starscope/scRNA-seq/vdj/reference/hg38_bcrtcr.fa"trust4_vdj_imgt_fasta="/starscope/scRNA-seq/vdj/reference/human_IMGT+C.fa"// strand reverse
soloStrand="Reverse"}// uncomment line below if using slurm, see https://www.nextflow.io/docs/latest/executor.html
// process.executor = 'slurm'
// uncomment below chunk if using conda
// process.conda = "/home/xzx/Tools/mambaforge/envs/starscope_scRNAseq_env"
// conda.enabled = true
// docker setting, comment chunk below if using conda
process.container="registry-intl.cn-hangzhou.aliyuncs.com/thunderbio/starscope_scrnaseq_env:1.2.5"docker.enabled=truedocker.userEmulation=truedocker.runOptions='--init-u$(id-u):$(id-g)$(opt="";forgroupin$(id-G);doopt=$opt" --group-add $group";done;echo$opt)'// Resouces for each process
process{withLabel:process_high{cpus=16memory=40.GB}withLabel:process_medium{cpus=4memory=20.GB}withLabel:process_low{cpus=4memory=20.GB}withName:CHECK_SATURATION{cpus=4memory=10.GB}withName:CAT_FASTQ{cpus=2memory=4.GB}withName:TRIM_FASTQ{cpus=12memory=20.GB}withName:MULTIQC{cpus=4memory=10.GB}withName:STARSOLO{cpus=16memory=40.GB}withName:REPORT{cpus=4memory=40.GB}withName:FEATURESTATS{cpus=2memory=8.GB}withName:GENECOVERAGE{cpus=8memory=10.GB}withName:VDJ_CELLCALLING_WITHOUTGEX{cpus=10memory=20.GB}withName:VDJ_CELLCALLING_WITHGEX{cpus=10memory=20.GB}withName:GET_VERSIONS_VDJ{cpus=2memory=10.GB}withName:VDJ_ASSEMBLY{cpus=32memory=30.GB}withName:VDJ_METRICS{cpus=2memory=10.GB}withName:REPORT_VDJ{cpus=4memory=40.GB}}
By default, the intermediate files will be written to sub-directory of
work under the pipeline running directory, please feel free to
remove it after all the processes finished successfully.
Chapter 4
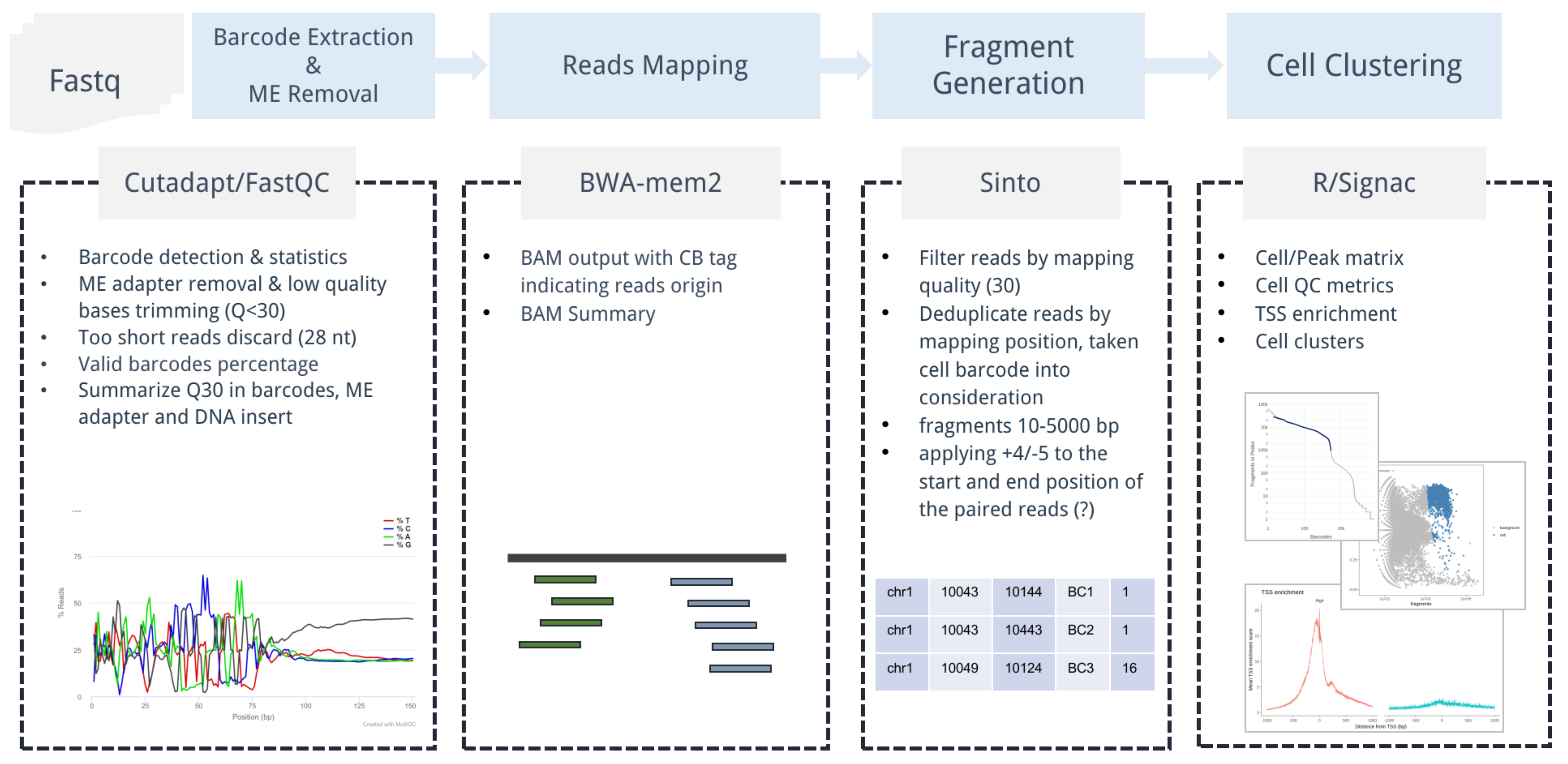
scATAC workflow
Subsections of scATAC workflow
Invoke with StarScope
Input
The input sample list file is a simple csv with three columns.
The first column sample indicates sample IDs, and multiple fastq
files with the same sample ID will be concatenated before further
processing (e.g. two pairs of human_pbmc_s1 fastq files will be
cat to a single pair). Multiple samples in one single sample list
will be submitted parallelly and processed asynchronously. The workflow
supports both ThunderBio and 10X ATAC data, please use the suggested
configuration below. User could check 10X ATAC library structure
from https://teichlab.github.io/scg_lib_structs/.
All samples in the sampleList will use the same options. Therefore, a combination of samples from different species is not supported.
Warning
We found that seq library doesn’t support fastq file with quality
score larger than 40 (denoted as J in the fourth line). Please check the quality encoding
on FASTQ wiki page. The error message is like:
User could use the command below to convert “J” (Q=41) to “I” (Q=40):
for i in *.fq.gz;do zcat $i| awk 'NR%4==1{print; getline; print; getline; print; getline; gsub("J", "I"); print}'| gzip -c > ${i%%.fq.gz}".modified.fq.gz";done
Command
Invoke with config file
Since there are too many processes and options, we recommend to invoke pipeline with custom
configuration file, which gives users more control on resources management.
params{bwaIndex="/refdata/human/bwa2_index/GRCh38-2020-A"genomeGTF="/refdata/human/refdata-gex-GRCh38-2020-A/genes/genes.gtf"whitelist="/barcodes/BC1.tsv /barcodes/BC2.tsv /barcodes/BC3.tsv"// TB whitelist
// trimming options used by cutadapt
trimOpt='-aAGATGTGTATAAGAGACAG...CTGTCTCTTATACACATCTCCGAGCCCACGAGAC-AGTCTCGTGGGCTCGGAGATGTGTATAAGAGACAG...CTGTCTCTTATACACATCT'// cell filtration
minCell=10// feature obeseved in at least "minCell" cells
minFeature=200// cells with at least "minFeature" features
publishSaturation=trueplatform="TB"// TB or 10X
}// uncomment line below if using slurm, see https://www.nextflow.io/docs/latest/executor.html
// process.executor = 'slurm'
// uncomment below chunk if using conda
// process.conda = "/home/xzx/Tools/mambaforge/envs/starscope_scATAC_env"
// conda.enabled = true
// docker setting, comment chunk below if using conda
process.container="registry-intl.cn-hangzhou.aliyuncs.com/thunderbio/starscope_scatac_env:0.0.6"docker.enabled=truedocker.userEmulation=truedocker.runOptions='--init-u$(id-u):$(id-g)$(forgroupin$(id-G);doecho"--group-add $group";done)'// Resouces for each process
process{withLabel:process_high{cpus=16memory=32.GB}withLabel:process_medium{cpus=16memory=20.GB}withLabel:process_low{cpus=16memory=20.GB}withName:CAT_FASTQ{cpus=2memory=10.GB}withName:CAT_FASTQ_10X{cpus=2memory=10.GB}withName:REPORT{cpus=8memory=40.GB}withName:CHECK_BARCODE{cpus=16memory=20.GB}CHECK_BARCODE_10X{cpus=16memory=20.GB}withName:TRIM_FASTQ{cpus=16memory=20.GB}withName:BWA_MAPPING{cpus=16memory=32.GB}withName:STATS{cpus=4memory=20.GB}withName:DEDUP{cpus=2memory=20.GB}withName:CHECK_SATURATION{cpus=4memory=20.GB}withName:MULTIQC{cpus=4memory=20.GB}withName:GENERATE_FRAGMENTS{cpus=16memory=40.GB}withName:SIGNAC{cpus=4memory=40.GB}}
user will have to provide --bwaIndex /thunderData/refdata/human/bwa2_index/GRCh38-2020-A to starscope command.
--genomeGTF: reference genome GTF file path.
--refGenome: reference genome assembly ID.
This parameter is used to extract genome size when calling peaks with macs2. Here we support
hg38, hg19, mm9 and mm10 for human and mosue genome. User could use --genomeSize to indicate
genome size directly.
--platform: StarScope scATAC pipeline supports ATAC data from both the ThunderBio and 10X genomics. Use either
TB or 10X here.
--whitelist: The white list file(s) path. For ThunerBio scATAC data, whitelist files were distributed with StarScope:
Each sample will have a separated result folder named with sample ID. The subdirectory final
contains all the result files, including html report, fragments file (with all the barcodes)
and a list of cell associated barcodes identified (e.g. TB_PBMC_s1_raw_cells.tsv).
The pipeline_info directory contains statistics of the pipeline running resources.
By default, the intermediate files will be written to subdirectory of
work under the pipeline running directory, please feel free to
remove it after all the processes finished successfully.